Structural Variant (SV) Genotyping Ongoing...
Structural variants (SVs) play a causal role in numerous diseases but are difficult to detect and accurately genotype (determine zygosity) in whole-genome short-read sequencing data. SV genotypers that assume that the aligned sequencing data uniformly reflect the underlying SV or use existing SV call sets as training data can only partially account for variant and sample-specific biases. We are developing novel genotypers that combined machine learning-methods and SRS simulation to improve SV genotyping accuracy.
The observed SRS data reflects the genome induced by a putative SV but also depends on interactions between the genomic region, sequencing process, and analysis software. Our key idea is to use SRS simulation to generate empirical distributions of the expected SV evidence instead of trying to build a model of those complex and interconnected effects. Our current tool, NPSV-deep, builds on NPSV’s (its predecessor) use of simulated training data, but recasts SV genotyping as an image similarity problem that can exploit deep learning approaches. We model SV genotyping as a facial recognition task, with many (potentially unbounded) outputs and few examples per output, instead of image classification, with its fixed number of outputs and many examples per output. NPSV-deep improves accuracy for SVs as currently described, and by leveraging the learned similarity model, enables automatic correction of imprecise/incorrect SV descriptions (that otherwise negatively impact genotyping accuracy).
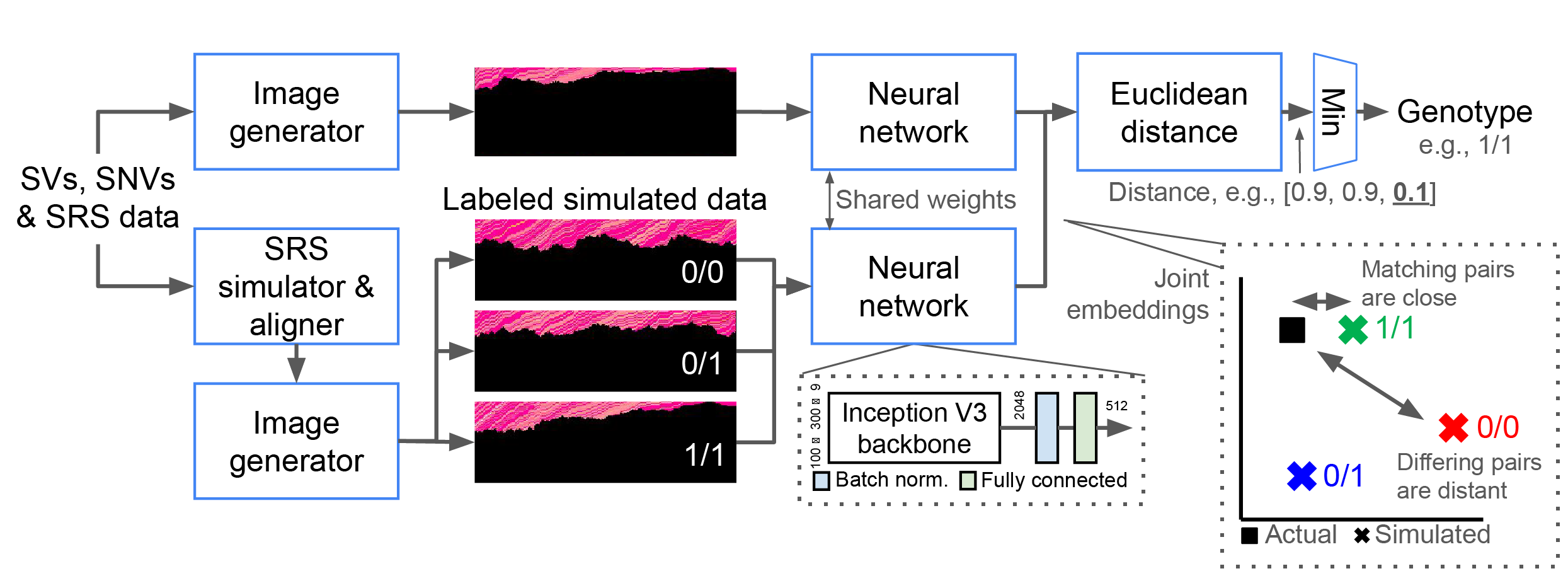 For each SV and possible genotype, NPSV-deep generates and aligns 1+ simulated replicates using a SRS simulator and pipeline configured to match the actual sequencing data. The genotype is determined from the closest pair of embeddings of the actual and labeled simulated data. Adapted from Linderman et al. Bioinformatics 2024.
For each SV and possible genotype, NPSV-deep generates and aligns 1+ simulated replicates using a SRS simulator and pipeline configured to match the actual sequencing data. The genotype is determined from the closest pair of embeddings of the actual and labeled simulated data. Adapted from Linderman et al. Bioinformatics 2024.
These efforts are described in the following publications:
- Linderman MD, Wallace J, van der Heyde A, Wieman E, Brey D, Shi Y, Hansen P, Shamsi Z, Gelb BD, Bashir A. NPSV-deep: A deep learning method for genotyping structural variants in short read genome sequencing data. Bioinformatics. 2024;40(3).
- Linderman MD, Paudyal C, Shakeel M, Kelley W, Bashir A, Gelb BD. NPSV: A simulation-driven approach to genotyping structural variants in whole-genome sequencing data. Gigascience. 2021;10(7).
PeopleSeq Ongoing...
PeopleSeq is a longitudinal study of healthy adults who plan to, or have already received, their own genomic sequence information. The PeopleSeq study is the collaborative effort of a growing consortium of commercial and research organizations. The PeopleSeq study will collect valuable empirical data on the medical, behavioral and economic impact of performing elective personal genome sequencing.
PeopleSeq is described in the following publications:
- Linderman MD, Adelson SM, Berro TM, Anderson JL, Crawford SD, Cunningham TJ, et al. Elective genomic sequencing for adults in research, clinical and commercial contexts. medRxiv 2026. To appear in BMC Med Genomics.
- Zoltick ES, Linderman MD, McGinniss MA, Ramos E, Ball MP, Church GM, et al. Predispositional genome sequencing in healthy adults: design, participant characteristics, and early outcomes of the PeopleSeq Consortium. Genome Med. 2019;11(1):10.
- Linderman MD, Nielsen DE, Green RC. Personal Genome Sequencing in Ostensibly Healthy Individuals and the PeopleSeq Consortium. J Pers Med. 2016;6(2):14.
Horizontally Scalable Genome Analysis Completed...
The growing generation of genomic data motivates the adoption of the scalable distributed computing systems already in wide use in other “big data” domains. ADAM is genome analysis platform built on Apache Avro, Apache Spark and Parquet. We have been experimenting with porting existing genomics tools to ADAM. Our Distributed Exome CNV Analyzer, or DECA, is a distributed re-implementation of the XHMM exome CNV caller using ADAM and Apache Spark.
These efforts are described in the following publications:
- Linderman MD, Chia D, Wallace F, Nothaft FA. DECA: scalable XHMM exome copy-number variant calling with ADAM and Apache Spark. BMC Bioinformatics. 2019;20:493.
- Nothaft FA, Massie M, Danford T, Zhang Z, Laserson U, Yeksigian C, et al. Rethinking Data-Intensive Science Using Scalable Analytics Systems. Proceedings of the 2015 ACM SIGMOD International Conference on Management of Data. Melbourne, Victoria, Australia: ACM; 2015. p. 631–46.
Genomics Education Completed...
There is an acute need to develop more effective genomics education to train the next-generation of genomics professionals. With a multi-disciplinary group of colleagues at the Icahn of School of Medicine at Mount Sinai, I co-developed and directed a laboratory-style medical genomics course (first taught in 2012), in which the students have the opportunity to sequence and analyze their own whole genome. The results of our companion research study evaluating student attitudes towards and the outcomes of incorporating personal genome sequencing into graduate genomics education are described in the following publications:
- Linderman MD, Sanderson SC, Bashir A, Diaz GA, Kasarskis A, Zinberg R, et al. Impacts of incorporating personal genome sequencing into graduate genomics education: a longitudinal study over three course years. BMC Med Genomics. 2018;11(1):5.
- Linderman MD, Bashir A, Diaz GA, Kasarskis A, Sanderson SC, Zinberg RE, et al. Preparing the next generation of genomicists: a laboratory-style course in medical genomics. BMC Med Genomics. 2015;8:47.
- Sanderson SC, Linderman MD, Zinberg R, Bashir A, Kasarskis A, Zweig M, Suckiel S, Shah H, Mahajan M, Diaz GA, Schadt EE. How Do Students React to Analyzing Their Own Genomes in a Whole-Genome Sequencing Course?: Outcomes of a Longitudinal Cohort Study. Genet Med. 2015;17: 866–74.
- Sanderson SC, Linderman MD, Kasarskis A, Bashir A, Diaz GA, Mahajan M, Shah H, Wasserstein M, Zinberg RE, Zweig M, Schadt EE. Informed decision-making among students analyzing their personal genomes on a whole genome sequencing course: a longitudinal cohort study. Genome Med. 2013 Dec 30;5(12):113.
To support hands-on genomics education, I developed the MySeq web application. MySeq is a web-based, privacy protecting, personal genome analysis tool inspired by GENOtation (previously the Interpretome) and DNA.LAND Compass. MySeq can load and analyze Tabix-indexed VCF files stored locally on the user’s computer or available remotely. Queries and other analyses will only load the necessary blocks of the compressed VCF file, enabling efficient analysis of whole-genome-scale VCF files. MySeq has been used in the CSCI1007 “Practical Analysis of a Personal Genome” winter-term course at Middlebury College. MySeq is described in the following publication:
- Linderman MD, McElroy L, Chang L. MySeq: privacy-protecting browser-based personal Genome analysis for genomics education and exploration. BMC Med Genomics. 2019;12(1):172.
Accurately assessing genomics knowledge was an ongoing challenge in this and other projects. In response, we developed and validated the GKnowM genomics knowledge scale. GKnowM incorporates modern genomics technologies and is informative for individuals with diverse backgrounds, including those with clinical/life-sciences training. We performed a rigorous psychometric evaluation of GKnowM with a large, educationally, and ethnically diverse cohort drawn from the general public, students, and genomics professionals. GKnowM is described in the following publications:
- Linderman MD, Suckiel SA, Thompson N, Weiss DJ, Roberts JS, Green RC. Development and Validation of a Comprehensive Genomics Knowledge Scale. Public Health Genomics. 2021;24(5–6):291–303.
HealthSeq Completed...
HealthSeq is a longitudinal cohort study at Mount Sinai in which unselected ostensibly healthy participants received a variety of health and non-health-related genetic results from whole genome sequencing. The primary aim of HealthSeq is to improve our understanding of participants’ motivations, expectations, concerns and preferences, and the impacts of receiving personal genome sequencing in a pre-dispositional setting.
HealthSeq results are described in the following publications:
- Sanderson SC, Linderman MD, Suckiel SA, Zinberg R, Wasserstein M, Kasarskis A, et al. Psychological and behavioural impact of returning personal results from whole-genome sequencing: the HealthSeq project. Eur J Hum Genet. 2017;25(3):280-92.
- Suckiel SA, Linderman MD, Sanderson SC, Diaz GA, Melissa Kasarskia AW, Schadt EE, et al. Impact of genomic counseling on informed decision-making among ostensibly healthy individuals seeking personal genome sequencing: The HealthSeq project. J Genet Couns. 2016;25(5):1044-1053.
- Sanderson SC, Linderman MD, Suckiel SA, Diaz GA, Zinberg RE, Ferryman K, et al. Motivations, concerns and preferences of personal genome sequencing research participants: Baseline findings from the HealthSeq project. Eur J Hum Genet. 2016 Jun 3;24(1):14–20.
Genome Analysis Pipeline Completed...
I developed the Genome Analysis Pipeline (GAP) used by many groups at the Icahn Institute for Genomics and Multiscale Biology at Mount Sinai. The GAP was validated for clinical use in New York State and has been successfully applied to identify causal mutations in multiple patients. The GAP is used in multiple small and large research projects, totalling many thousands genomes, exomes and targeted panels.
The GAP is described in the following publications:
- Linderman MD, Brandt T, Edelmann L, Jabado O, Kasai Y, Kornreich R, Mahajan M, Shah H, Kasarskis A, Schadt EE Analytical Validation of Whole Exome and Whole Genome Sequencing for Clinical Applications. BMC medical genomics. 2014 Apr;7:20.
Large-scale Co-expression Analysis Completed...
Weighted Gene Co-expression Network Analysis (WGCNA) is a methodology for describing the correlation patterns among genes across microarray samples. Analysis of tens of thousands of probes, however, can take hours and requires hundreds of gigabytes of memory, putting this method out of reach for all but a few organizations and applications. Coexpp substantially reduces in the execution time and memory footprint. Those reductions will enable to researchers to apply WGCNA in many new contexts.
CytoSPADE Completed...
Recent advances in flow cytometry enable simultaneous single-cell measurement of 30+ surface and intracellular proteins. In a single experiment we can now measure enough markers to identify and compare functional immune activities across nearly all cell types in the human hematopoietic lineage. However, practical approaches to analyze and visualize data at this scale are only now becoming available. SPADE, described in Qiu et al., Nature Biotechnology 2011 and first used in Bendall et al. Science 2011, is a novel algorithm that organizes cells into hierarchies of related phenotypes, or “trees”, that facilitate the visualization of developmental lineages, identification of rare cell types, and comparison of functional markers across stimuli.
CytoSPADE is a robust, modular and performant implementation of Qiu et al.’s SPADE algorithm, including a rich GUI implemented as a plugin for the Cytoscape Network Visualization platform. CytoSPADE is 12-19 fold faster than the SPADE prototype, ensuring that users can run complex analyses on their laptops in just seconds or minutes. More information is available on the software page.
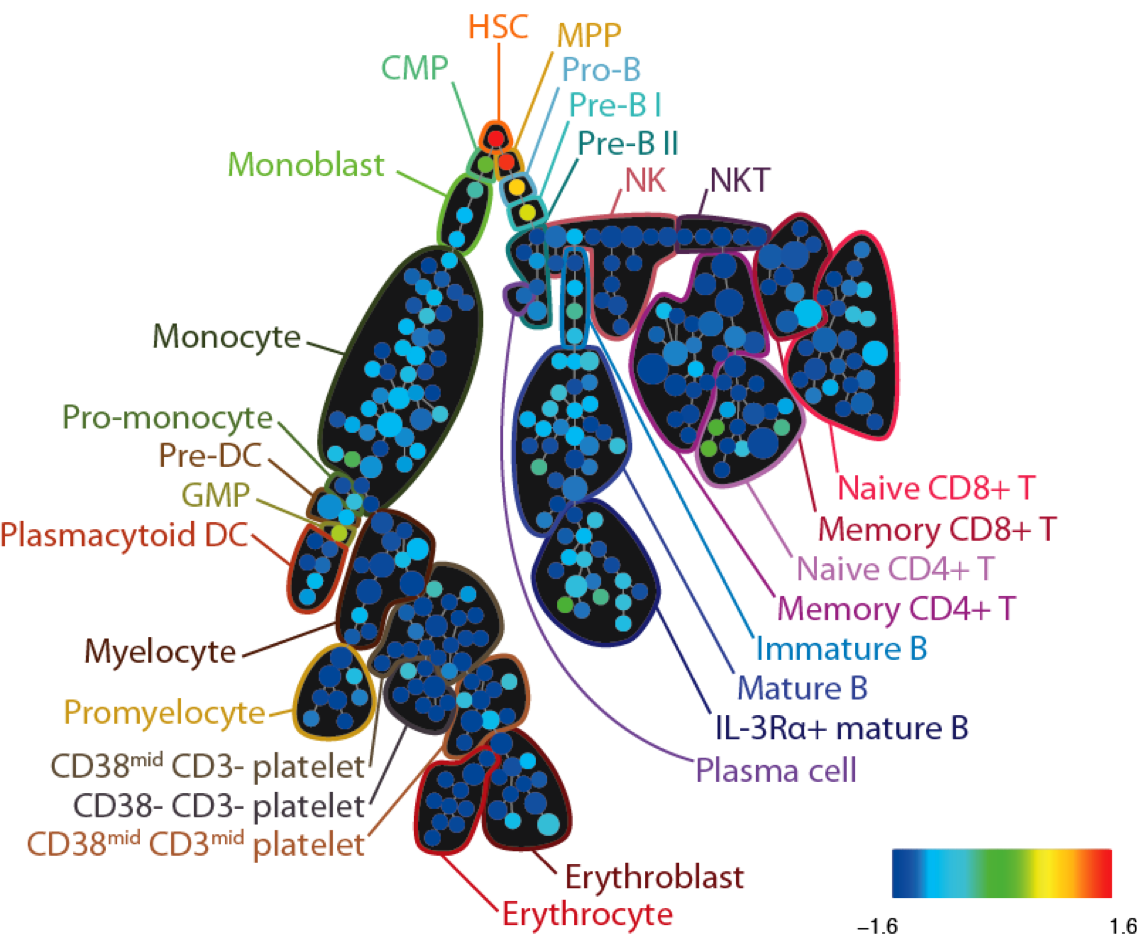 SPADE tree annotated with CD34 expression in healthy human bone marrow (PBMC) samples analyzed via mass cytometry. Adapted from Bendall et al. Science 2011.
SPADE tree annotated with CD34 expression in healthy human bone marrow (PBMC) samples analyzed via mass cytometry. Adapted from Bendall et al. Science 2011.
CytoSPADE is described in the following publications:
- Anchang B, Hart TDP, Bendall SC, Qiu P, Bjornson Z, Linderman M, et al. Visualization and cellular hierarchy inference of single-cell data using SPADE. Nat Protoc. 2016;11(7):1264–79.
- Linderman MD, Bjornson Z, Simonds EF, Qiu P, Bruggner R, Sheode K, Meng TH, Plevritis SK, Nolan GP. CytoSPADE: High-Performance Analysis and Visualization of High-Dimensional Cytometry Data. Bioinformatics. 2012;15(18):2400-1.
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr., Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29(10):886-91.
Bayesian Structure Learning Completed...
Aberrant intracellular signaling plays an important role in many diseases. The causal structure of signal transduction networks can be modeled as Bayesian Networks (BNs), and computationally learned from experimental data. However, learning the structure of BNs is an NP-hard problem that, even with fast heuristics, is too time consuming for large, clinically important networks (20-50 nodes). I developed a novel graphics processing unit (GPU)-accelerated implementation of a Monte Carlo Markov Chain-based algorithm for learning BNs that is up to 7.5-fold faster than already heavily optimized general-purpose processor (GPP)-based implementations.
The GPU-based implementation is just one of several variants within the larger application, each optimized for a different input or machine configuration. I concurrently enhanced the Merge framework to enable efficient integration, testing and intelligently selection among the different potential implementations targeting multicore GPPs, GPUs and distributed compute clusters.
The GPU-accelerated Bayesian Network learning implementation is described in the following publication:
- Linderman MD, Bruggner R, Athalye V, Meng TH, Asadi NB, Nolan GP. High-throughput Bayesian network learning using heterogeneous multicore computers. Proc ACM Intl Conf on Supercomputing (ICS); 2010. p. 95-104.
The performance of the BN learning application is sensitive to the performance of of an accumulation of log-space probabilities in the inner most loop:
acc += log(1 + exp(x))
For x far from the origin, this computation can be approximated as 0 or the
identity function. The choice of those boundaries creates a
performance-precision trade-off. I concurrently developed a tool, Gappa++, for
analyzing the numerical behavior of this and other computations. Using Gappa++
I as able to improve performance an additional 10-15%. Gappa++ is described in
the following publication and on the software page:
- Linderman MD, Ho M, Dill DL, Meng TH, Nolan GP. Towards program optimization through automated analysis of numerical precision. Proc IEEE/ACM Intl Symp on Code Generation and Optimization (CGO); 2010. p. 230-7.
Merge Completed...
Computer systems are undergoing significant change: to improve performance and efficiency, architects are exposing more microarchitectural details directly to programmers. Software that exploits specialized accelerators, such as GPUs, and specialized processor features, such as software-controlled memory, exposes limitations in existing compiler and OS infrastructure.
Merge is a programming model for building applications that will tolerate changing hardware. Merge allows programmers to leverage different processor-specific or domain-specific toolchains to create software modules specialized for different hardware configurations, and it provides language mechanisms to enable the automatic mapping of the application to these processor-specific modules. I showed that this approach can be used to manage computing resources in complex heterogeneous processors and to enable aggressive compiler optimizations.
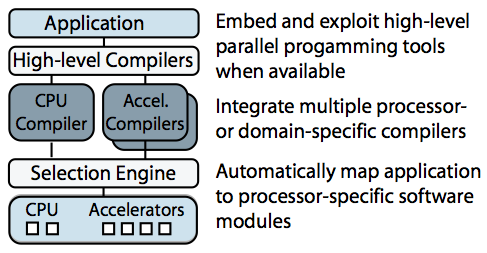 Sketch of the Merge framework
Sketch of the Merge framework
Merge was used extensively if the complex and computationally intensive Bayesian structure learning application described above. Using Merge we were able to deploy a single application binary that could deliver the best possible performance across a range of problem sizes and hardware configurations (including multicore processors, GPUs and clusters with MPI). For any given problem size and hardware configuration, the Merge-enabled application automatically and dynamically selects the appropriate implementation (based on predicates supplied by original implementors).
Merge is described in the following publications:
- Linderman MD, Balfour J, Meng TH, Dally WJ. Embracing heterogeneity: parallel programming for changing hardware. Proc USENIX Conf on Hot Topics in Parallelism (HOTPAR); 2009.
- Linderman MD, Collins JD, Wang H, Meng TH. Merge: a programming model for heterogeneous multi-core systems. Proc Intl Conf on Architectural Support for Programming Languages and Operating Systems (ASPLOS); 2008. p. 287-96.