Test Project 1
Initial Due Date: 2024-10-24 8:00AM
Final Due Date: 2024-11-07 4:15PM
Background
A test project is an assignment that you complete on your own, without the help of others. It is like a take-home exam. You may use the textbook, your notes, your previous assignments, the notes and examples on the course web page and the Python library documentation (linked from the course web page), but use of any other source, such as Google, is not permitted. You may not discuss this assignment with anyone except the course instructor, ASIs and peer drop-in course assistants. However, all of those parties may only give you limited answers (as you would expect during an exam).
To be clear, you may not work with, discuss, or in any way collaborate with anyone else.
Try to get as far as you can. If you are stuck, contact me. While I cannot help you solve the test project itself, I may be able to suggest general problem-solving strategies, help with conceptual difficulties about Python, and or direct you to relevant examples from class.
Make sure you attempt all of the functions, even if you weren’t able to complete one of the “upstream” functions (i.e. make sure to attempt gene_finder even if you weren’t able to complete the find_orfs function it uses). Gradescope uses “upstream” functions from the solution to test your “downstream” functions and so your implementation for gene_finder can pass the tests even if your implementation of find_orfs does not.
Gene Finding
For this test project you will be writing a program to find candidate genes in the genome. No biology knowledge is required (although I hope you will learn a little about genomics along the way!). To help with some of the vocabulary there is a glossary at the end.
Proteins carry out the “work” of our cells and genes (in our DNA) contain the instructions for creating those proteins. In the central dogma of molecular biology, the DNA of a gene is transcribed into messenger RNA, which in turn is translated into the amino acids that make up a protein. This translation is performed in groups of three nucleotides termed codons. Thus the protein coding portion of DNA (i.e. a gene) consists of a sequence of codons. This sequence begins with the “ATG” start codon and ends at one of three stop codons (“TGA”, “TAG” or “TAA”). The start codon determines the reading frame, that is where to start “counting off” by threes. The DNA sequence from the beginning of the start codon up to but not including the next in-frame stop codon is called an open reading frame or ORF.
An example ORF is shown below in bold, with the codons in the ORF underlined.
ACATGATTCCCGGCTAAGTG
--- ---
--- ---
Your program will find candidate genes in a sequence of DNA, that is find ORFs in a string of capital ACGTs. You will do so by finding potential start codons in the sequence, and then for each start codon, scanning the DNA until you find a stop codon (e.g. “TAA” above). One subtlety is that DNA is double stranded. Genes can occur on either strand and thus you will need to scan both the DNA sequence and the other strand (its reverse complement) for genes. Each step of this process is described in more detail in the Guide below.
Specifications
Your program must include the following functions:
is_stop, which takes a string as an argument (i.e. a codon) and returns True if that string is a stop codon, i.e. if that string is “TGA”, “TAG” or “TAA”, and False otherwise.orf_sequence, which takes a DNA string as a parameter, and returns a string containing the ORF sequence. The string parameter will always start with the start codon, i.e. start with “ATG”.find_orfs, which takes a DNA string as a parameter and returns a list of strings that are ORFs in that DNA sequence. If no ORFs are found it should return an empty list.reverse_complement, which takes a DNA string as a parameter and returns the reverse complement of that DNA sequence as a string.gene_finder, which takes a DNA string as a parameter and returns a list of strings that are valid ORFs in that DNA sequence and its reverse complement.
To achieve the Exemplary assessment, use those functions along the functions provided in the skeleton code to identify candidate genes in the Salmonella genome (as described in the Guide). You will upload a file containing your predicted gene sequences to Gradescope along with your program.
You can assume that all DNA strings provided as arguments to your functions will only contain upper case letters.
Note: There is no “Creativity Points” component to this test project.
Guide
The required functions represent an approach to decomposing the large “gene finder” problem into smaller, more manageable, pieces. I encourage you to implement the functions in the order specified above, testing extensively as you go. Get started by downloading the starter file.
is_stop
is_stop should return True if its argument is a stop codon, i.e. one of “TGA”, “TAG” or “TAA”, and False otherwise. For example:
>>> is_stop("TAA")
True
>>> is_stop("TGA")
True
>>> is_stop("TAG")
True
>>> is_stop("ATG")
FalseTo facilitate implementing the remaining functions, is_stop should also return False (and not raise an error) if its argument is not a valid codon. For example:
>>> is_stop("TA")
Falseorf_sequence
orf_sequence will extract and return the ORF from a DNA string that begins with the start codon. That is orf_sequence will assume that its argument always begins with a start codon, i.e., always begins with “ATG”. The ORF is defined as the string beginning with the start codon up to but not including the in-frame stop codon, or if no stop codon is found, the end of the string.
Recall that codons are groups of three DNA nucleotides or bases (letters), thus “in-frame” is a position in the DNA string that is a multiple of 3 from the beginning of the start codon. For example the DNA sequence ATGAATAACTAGGC begins with the start codon “ATG”. The next codon will be “AAT”, followed by “AAC”, followed the stop codon “TAG”. The ORF for this sequence would be “ATGAATAAC”. It may look like there is an earlier stop codon “TAA” (at index 5), and thus a different ORF “ATGAA”, however that sequence is not a valid ORF because the “TAA” is not in-frame (i.e. not a multiple of 3 DNA bases from start codon).
Here are some examples of orf_sequence in use:
>>> orf_sequence("ATGAATAACTAGGC")
'ATGAATAAC'
>>> orf_sequence("ATGTAA")
'ATG'
>>> orf_sequence("ATGTAAATGAATAA")
'ATG'
>>> orf_sequence("ATGATAAAGGC")
'ATGATAAAGGC'Notice that in the last example there is no stop codon and so orf_sequence returns its entire argument.
The orf_sequence function should be relatively short. I suggest using a for loop (recall that range can take three parameters, an inclusive start, an exclusive end and a step) and the is_stop function you wrote previously. Note that while it is an error to index beyond the end of a sequence (e.g., a string), it is not an error to slice beyond the end of sequence (it will just evaluate to the end of the sequence). For example,
>>> "abc"[:100]
'abc'thus it is OK to try to slice 3 letters off the end of a string with fewer than 3 characters remaining, you will just get a shorter string, for example:
>>> i = 1
>>> "abc"[i:i+3]
'bc'find_orfs
find_orfs will return a list of ORFs in a DNA string assuming a single reading frame. That is find_orfs will scan a string from beginning to end in multiples of 3 bases. When a start codon is found, it will extract the ORF beginning at the start codon (using orf_sequence) and then resume scanning for a start codon immediately after the just extracted ORF. For example:
>>> find_orfs("AATGCCATGTGAATGCCCTAA")
['ATG', 'ATGCCC']These ORFs begin at indices 6 and 12 respectively. After finding the first ORF extending from index 6 (to index 11), find_orfs began scanning again at index 12, i.e., is skips ahead the length of string returned by orf_sequence (“ATG”) plus 3 for the stop codon. find_orfs did not report the “ATG” at index 1 because that is not in-frame, i.e. it is not a multiple of 3 bases from the beginning of the string.
In the following example notice that find_orfs skips the second “ATG” (i.e. the smaller nested ORF). That is the specified behavior as we are trying to identify larger ORFs.
>>> find_orfs("ATGCCCATGGGGAAATTTTGACCC")
['ATGCCCATGGGGAAATTT']Since both the number of ORFs and the amount you might need to “skip ahead” in the string is not known ahead of time, I suggest using a while loop. Recall that in a previous set of practice problems we implemented a while loop that printed all numbers from 1 to 20 inclusive that were divisible by 3, e.g.
i = 3
while i <= 20:
print(i)
i += 3In the above code the loop variable increments the same amount in each iteration. However it does not need to be that way, in each iteration the loop variable could change by a different amount (as long the loop eventually terminates). For example, the code below increments i by different amounts if i is even or odd. We could also increment i by an amount determined dynamically. The code below increments i by the length of the string representation of i if i is odd.
i = 3
while i <= 20:
print(i)
if i % 2 == 0:
i += 3
else:
i += len(str(i))reverse_complement
DNA is double stranded. The strands are complements of each other, i.e. the “A” on one strand is paired with a “T” on the other strand, “C” with a “G”, “G” with a “C” and “T” with an “A”.
Genes can be found on either strand, that is read from left to right (more formally 5’ to 3’) on the top strand or right to left (also 5’ to 3’) on the bottom strand. In the following example notice the two ORFs (in bold), one on the top strand the other on the bottom strand.
5' AATGCCGTGCTTGTAGACGTTACGACGTCATG 3' 3' TTACGGCACGAACATCTGCAATGCTGCAGTAC 5'
Our find_orfs function is written assuming its argument string should be read left to right (5’ to 3’) - like the top strand. To find ORFs on the other strand using find_orfs we need to convert the bottom strand to a string that can be read left to right i.e. “CATGACGTCGTAACGTCTACAAGCACGGCATT”. Doing so requires both reversing the order of and complementing the bases in the original DNA string. Your reverse_complement function should perform this transformation, e.g.
>>> reverse_complement("AATGCCGTGCTTGTAGACGTTACGACGTCATG")
'CATGACGTCGTAACGTCTACAAGCACGGCATT'
>> reverse_complement("GGAACCTT")
'AAGGTTCC'The order of the transformations does not matter. You can reverse the string first then complement the bases (i.e. convert the “A”s to “T”s, “G”s to “C”s, “C”s to “G”s, and “T”s to “A”s) or complement then reverse.
Recall that in a previous class we saw example implementations for reversing a string.
gene_finder
Now you should have all the pieces needed to implement a comprehensive gene finder. Your gene_finder function should take a DNA string as its argument and return a list with the all ORFs found for all three valid reading frames on both strands. Since each codon is 3 bases, there are three possible reading frames: all multiples of three starting at the first base, the second base or third base. That is for the DNA sequence “AATGCCATGTGAA”, your gene finder would identify all ORFs (using find_orfs) in the following six strings (the first three correspond to the “top” strand, and the second three correspond to the “bottom” or reverse complement strand):
"AATGCCATGTGAA"
"ATGCCATGTGAA"
"TGCCATGTGAA"
"TTCACATGGCATT"
"TCACATGGCATT"
"CACATGGCATT"Here is gene_finder in action with this DNA string as its argument:
>>> gene_finder("AATGCCATGTGAA")
['ATG', 'ATGCCATGTGAA', 'ATGGCATT']If we try to append a list, i.e., the list returned by find_orfs to a list we will get nested lists. To add all elements of one list to another we can use +=, which use the + concatentation operator for lists or the extend method.
Applying your gene finder to real data
While our functions may seem like they are not “real” tools, we can actually use our program to find real genes in a real genome. In this last part of the assignment, you will use your gene finder to identify candidate genes in a portion of the Salmonella genome.
The genomic sequence of interest is 6387 nucleotides long. When we run our gene finder on that genomic sequence we will identify more than 100 candidate ORFs. Most of these aren’t real genes, they are just ORFs that occur randomly by chance. Our hypothesis is that any real genes would be longer than the randomly occurring ORFs. Thus we want to filter by ORF length. We can set a threshold for that filter by generating random DNA sequences (that shouldn’t contain real genes), running our gene finder on those random sequences to generate candidate ORFs and finding the longest such ORF. We will use the length of the longest candidate ORF as our cutoff and only examine candidate ORFs longer than that threshold. I have already completed this process for you. Using 50 trials, I computed a cutoff of 690 nucleotides.
Using your functions and the functions included in the starter code, do the following:
- Download the text file containing a portion of the Salmonella genome and save it in the same folder as your program. For context, I obtained this genomic sequence, with accession number X73525, from GenBank, the NIH repository of genome sequences.
- Use the provided
read_fastafunction to read this file into a DNA string. - Use your
gene_finderfunction to compute a list of candidate ORFs in the DNA string you just read from the file. - Use the provided
filter_orfsfunction to create a new filtered list containing only ORFs longer than 690 nucleotides. Your filtered list should contain 5 candidate ORFs (including the ORF shown below). - Use the provided
write_fastafunction to save your ORFs as a text file named “genes.txt” that can be uploaded to Gradescope.
For example, applying your gene finder might look like:
sequence = read_fasta("X73525.fasta.txt")
orfs = gene_finder(sequence)
filtered_orfs = filter_orfs(orfs, 690)
write_fasta("genes.txt", filtered_orfs)You can perform the above operations in the Shell. Or if you want to complete these steps as part of your program, place the code inside the if __name__ == "__main__" statement included at the end of the starter file.
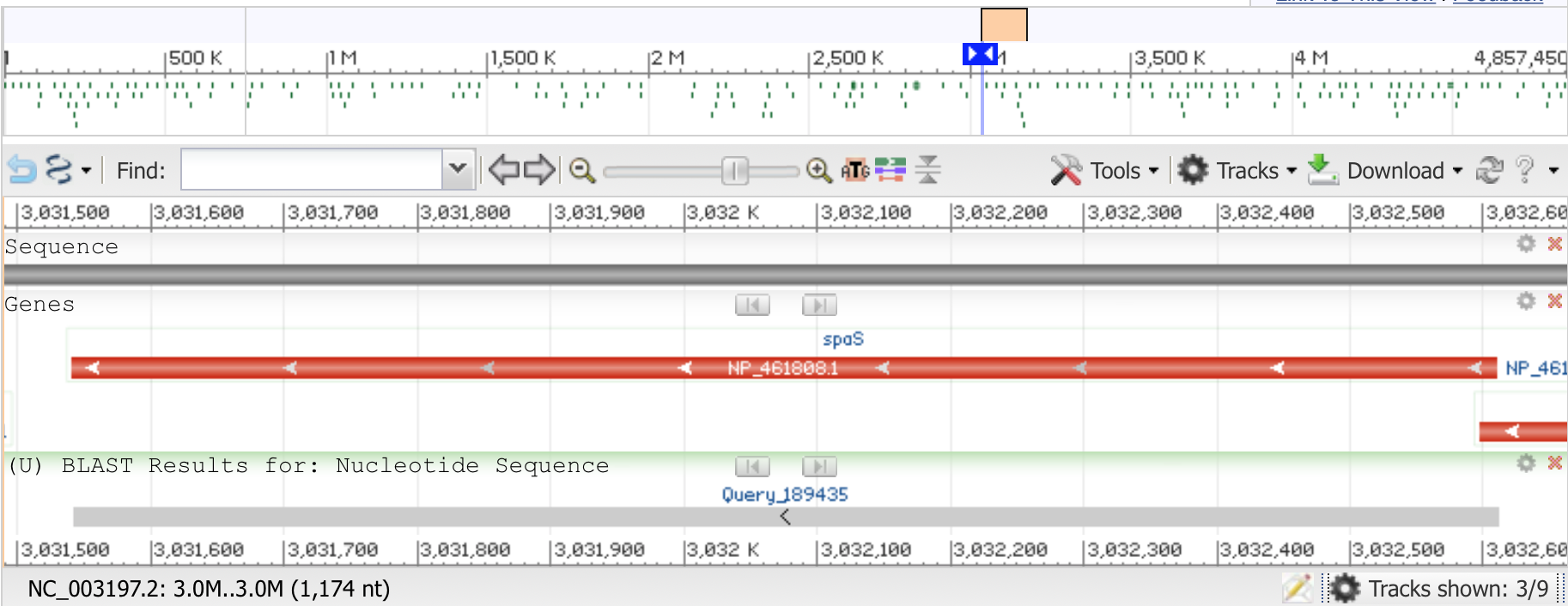
For your interest, I took one of the candidate ORFs I found in the Salmonella genome (copied below), and aligned it to the annotated Salmonella genome using the online BLAST tool. BLAST enables us to figure out where in a larger sequence(s), such as the entire Salmonella genome, a shorter sequence likely originated.
ATGTCCTCGAATAAAACAGAAAAACCGACTAAAAAACGGCTGGAAGACTCCGCTAAAAAAGGCCAGTCATTTAAAAGTAAAGATCTCATTATCGCCTGCCTGACGCTGGGAGGAATTGCCTATCTGGTGTCGTATGGCTCATTTAATGAGTTTATGGGGATAATTAAGATCATTATTGCGGATAATTTTGATCAGAGCATGGCTGACTACAGTTTGGCCGTTTTTGGGATAGGGTTAAAATATCTGATTCCATTTATGCTGCTCTGCTTAGTGTGTTCCGCATTACCGGCGTTATTACAGGCCGGTTTTGTGCTGGCGACAGAAGCATTAAAGCCTAATTTATCGGCGTTAAACCCGGTAGAAGGGGCAAAAAAACTTTTTAGTATGCGCACGGTTAAAGATACGGTCAAAACCCTACTGTATCTCTCATCCTTTGTGGTGGCCGCCATCATTTGCTGGAAGAAATATAAGGTTGAAATCTTTTCTCAGCTAAATGGCAATATTGTAGGTATTGCCGTCATTTGGCGTGAACTTCTCCTCGCATTGGTATTAACTTGCCTTGCTTGCGCATTGATTGTCTTATTATTGGATGCTATTGCGGAATATTTCCTGACCATGAAAGATATGAAAATGGATAAGGAAGAAGTGAAGCGTGAAATGAAGGAGCAGGAAGGGAACCCAGAGGTTAAATCTAAAAGACGTGAAGTTCATATGGAAATTCTGTCTGAACAGGTGAAATCTGATATTGAAAACTCACGCCTGATTGTTGCCAACCCCACGCATATTACGATCGGGATTTATTTTAAACCCGAATTGATGCCGATTCCGATGATCTCGGTGTATGAAACGAATCAGCGCGCACTGGCCGTCCGCGCCTATGCGGAGAAGGTTGGCGTACCTGTGATCGTCGATATCAAACTGGCGCGCAGTCTTTTCAAAACCCATCGCCGTTATGATCTGGTGAGTCTGGAAGAAATTGATGAAGTTTTACGTCTTCTGGTTTGGCTGGAAGAGGTAGAAAACGCGGGCAAAGACGTTATTCAGCCACAAGAAAACGAGGTACGGCATHere is a screenshot of the BLAST output. The “x-axis” is the position in the Salmonella genome. The candidate ORF identified with our program is shown in the “BLAST Results” section in the image below. A known gene in the Salmonella genome is shown as the red bar in the “Genes” section. Notice how closely our prediction lines up with the known gene! Your test project is a real gene finder!
When you’re done
Make sure that your program is properly documented:
- You should have a comment at the very beginning of the file with your name, and section.
- Each function should have an appropriate docstring (including arguments and return value if applicable).
- Other miscellaneous inline/block comments if the code might otherwise be unclear.
In addition, make sure that you’ve used good coding style:
- Use constants where appropriate
- Avoid code duplication as much as possible and define functions to implement repeated computations.
- Avoid global variables, i.e., variables used inside a function that were declared outside of the function (except for constants). Use parameters instead.
- Use meaningful names for your functions, variables, and parameters.
- Make your code readable by avoiding long lines that wrap, and using horizontal and vertical whitespace as appropriate.
Submit your program and the “genes.txt” file via Gradescope. You must submit your program and the “genes.txt” file at the same time (you can select multiple files for upload). Your program file must be named tp1_genes.py. You can submit multiple times, with only the most recent submission (before the due date) graded. Passing all of the visible tests does not guarantee that your submission correctly satisfies all of the requirements of the assignment.
Grading
| Assessment | Requirements |
|---|---|
| Revision needed | Some but not all tests are passing. |
| Meets Expectations | All tests pass, the required functions are implemented correctly and your implementation uses satisfactory style. |
| Exemplary | All requirements for Meets Expectations, you correctly identify the genes in the real data (i.e., genes.txt is correct), and your implementation is clear, concise, readily understood, and maintainable. |
Glossary
- DNA string
- A string composed of the characters ‘A’, ‘C’, ‘G’, ‘T’. For this assignment, we can assume the letters are always uppercase.
- codon
- A DNA string of length 3; i.e., a 3-letter string of ACGTs
- start codon
- The string “ATG”, which starts the DNA sequence for a gene
- stop codon
- Any of the codons “TGA”, “TAG”, or “TAA”, which indicate the end of the gene sequence
- ORF or ORF sequence
- An Open Reading Frame or ORF is a DNA string that can be translated to a protein. In this context, an ORF is a string starting with “ATG”; it extends up to but not including a stop codon (reading the string in groups of 3 letters), or if no stop codon is found, up to the end of the string. For example, the string “ATGAATTAGGC” starts with “ATG” and includes the stop codon “TAG” starting at position 6, a multiple of 3 from the beginning of the string, so the ORF would be “ATGAAT”. For the string “ATGATAGGC” the entire string “ATGATAGGC” would be the ORF because no stop codon is in a position a multiple of 3 away from the start.
- reverse complement
- The sequence of the other strand of DNA. To generate the reverse complement sequence, reverse the DNA string, and replace ‘A’ with ‘T’; ‘C’ with ‘G’; ‘G’ with ‘C’; and ‘T’ with ‘A’. For example, given string “ATG” the reverse is “GTA”, and then doing the substitutions we get “CAT”.
- base or nucleotide
- One of the letters ‘A’, ‘C’, ‘G’, or ‘T’
- reading frame
- A multiple of 3 characters. Any string will have 3 possible reading frames.
Adapted from Libeskind-Hadas, R., and Bush, E.(Libeskind-Hadas and Bush 2014)